Despite recent approvals of new agents and declining mortality in many high-income countries,
metastatic castration-resistant prostate cancer (mCRPC) remains to be lethal and there is a high
medical need for novel therapeutic strategies. To date, the standard of care for previously untreated
patients with mCRPC comprises androgen receptor pathway inhibitors (ARPI) together with androgen deprivation therapy (ADT).1 Furthermore, inhibition of poly (adenosine diphosphate [ADP]) ribose polymerase (PARP) has been reported to increase the activity of ARPI via an androgen receptor (AR)-dependent transcription. Vice versa, ARPI induces homologous recombination repair (HRR) deficiency, increasing the susceptibility to PARP inhibitors (PARPi).2 As such, co-inhibition of the AR signalling pathway and PARP might result in enhanced benefit with the potential to extend efficacy of PARPi to tumours regardless of HRR gene alterations.1,2
For patients with prostate cancer, there is a strong biological rationale to combine a PARPi with ARPI. First of all, AR inhibition suppresses the expression of HRR genes, enhancing sensitivity to PARPi. Secondly, PARP inhibition may reduce AR transcriptional activity, hence increasing sensitivity to ARPI. Finally, some cases of metastatic prostate cancer show a more aggressive disease phenotype and a weaker response to ARPI due to the loss of the RB transcriptional corepressor 1 (RB1) gene, which is often co-deleted with BRCA2, justifying the addition of a PARPi to treatment with ARPI in this subset of patients. Together, these three rationales support a potential synergistic effect of the ARPI-PARPi combination, regardless of HRR status, and have led to the initiation of clinical trials – and subsequent EMA-approval – of three ARPI-PARPi combinations in frontline mCRPC.2
In the phase III PROpel study, first-line mCRPC patients, regardless of HRR status, were randomised to olaparib and abiraterone versus placebo and abiraterone. All patients also received prednisone or prednisolone. The primary endpoint of investigator-assessed imaging-based (ib) PFS was met with a significantly longer ibPFS in the olaparib plus abiraterone arm than in the placebo and abiraterone arm (24.8 vs. 16.6 months, HR[95%CI]:0.66[0.54-0.81], p< 0.001).3 Furthermore, median overall survival (OS) was 42.1 months with olaparib plus abiraterone and 34.7 months with placebo plus abiraterone (HR[95%CI]: 0.81[0.67-1.00], p= 0.054).4
In the phase III MAGNITUDE study, mCRPC patients were randomised to first-line treatment with niraparib and abiraterone-prednisone versus placebo and abiraterone-prednisone. The primary endpoint of this study was radiographic PFS assessed by blinded independent central review (BICR). In the BRCA1/2 subgroup, niraparib plus abiraterone significantly improved rPFS (19.5 vs. 10.9 months, HR[95%CI]:0.55[0.39-0.78], p=0.0007). rPFS was also prolonged in the total HRR mutated population (16.7 vs. 13.7 months, HR[95%CI]: 0.76[0.60-0.97], p=0.0280).5 However, in nonmutated HRR patients, combination therapy did not result in a benefit in the composite endpoint of time to PSA progression and/or rPFS (HR[95%CI]: 1.09[0.75-1.57], p= 0.66).6 Finally, EMA-approval of the talazoparib-enzalutamide
combination was based on the results of the TALAPRO-2 study.1
The phase III TALAPRO-2 study aimed to investigate the combined efficacy of talazoparib (0.5 mg/day) plus enzalutamide (160 mg/day) as a first-line treatment in men with mCRPC. Patients unselected for genetic alterations in DNA damage repair pathways, directly or indirectly involved with HRR, received either talazoparib plus enzalutamide or placebo plus enzalutamide as first-line treatment for mCRPC. Patients were stratified by prior abiraterone or docetaxel for castration-sensitive prostate cancer and HRR gene alteration status. In order to be eligible for the trial, patients must have asymptomatic or mildly symptomatic mCRPC with disease progression at study entry, an ECOG performance status score ≤1, ongoing ADT and no prior life-prolonging therapy for CRPC. Previous docetaxel and abiraterone or orteronel in the castration-sensitive setting were allowed. Primary endpoint of the trial is rPFS by BICR. In total, 402 patients were randomised to talazoparib plus enzalutamide and 403 patients to placebo plus enzalutamide. Baseline demographics and disease characteristics were well-balanced between treatment arms. All patients had prospective tumour tissue test results; 21% had HRR gene alterations detected (HRR-deficient population) and 79% had no HRR gene alterations detected (HRR non-deficient population) or were of unknown alteration status. BRCA alterations were detected in 7% of patients in the talazoparib group and in 8% of patients in the placebo group.1
After a median follow-up of 24.9 months in the talazoparib arm and of 24.6 months in the placebo arm, treatment with talazoparib plus enzalutamide resulted in a 37% reduction in the risk of radiographic progression or death (HR[95%CI]: 0.63[0.51-0.78], p< 0.0001). Median rPFS was not reached for the talazoparib group and was 21.9 months for the placebo group (Figure 1). In subgroup analyses by HRR
gene alteration status, rPFS was also significantly improved in HRR-deficient (HR[95%CI]: 0.46[0.30–0.70], p= 0.0003), HRR-non-deficient or unknown (HR[95%CI]: 0.70[0.54–0.89], p= 0.0039), and HRR-non-deficient patients by prospective tumour tissue testing (HR[95%CI]: 0.66[0.49–0.91], p= 0.0092) in the talazoparib vs. placebo arm.1
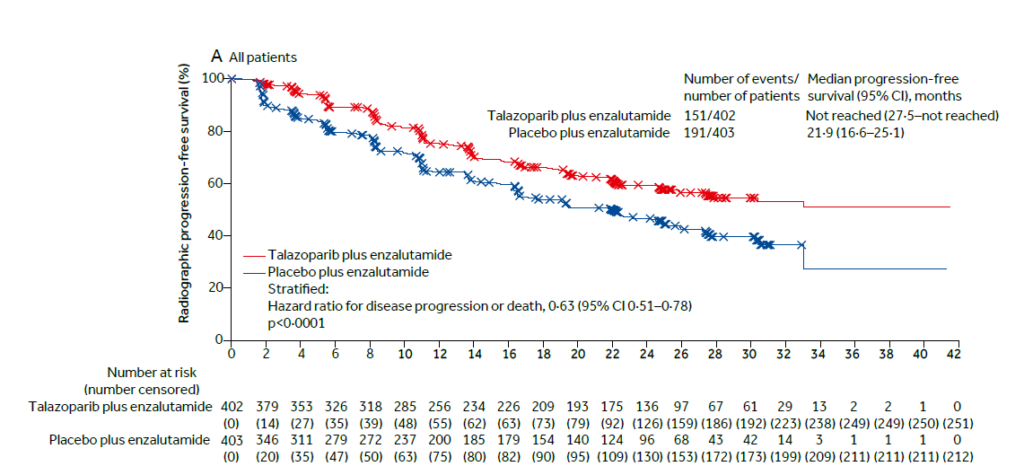
Figure 1. Radiographic progression-free survival in all patients.
A consistent treatment effect with talazoparib plus enzalutamide was seen in prespecified subgroups, including age, ECOG performance status, Gleason score, stage at diagnosis, site of metastasis and prior treatment with abiraterone or docetaxel. Overall survival (OS) data are still immature (31% maturity) but demonstrate a trend in favour of the talazoparib plus enzalutamide combination (HR[95%CI]: 0.89[0.69-1.14], p= 0.35). Furthermore, objective response rates were 62% for talazoparib plus enzalutamide
and 44% for placebo plus enzalutamide, with complete responses in respectively 38% and 18% of patients. Finally, the addition of talazoparib significantly prolonged time to PSA progression (p= 0.002), time to cytotoxic chemotherapy initiation (p< 0.0001) and time to progression or death on first
subsequent antineoplastic therapy (p= 0.036).1
Overall, 59% (talazoparib plus enzalutamide) and 18% (placebo plus enzalutamide) of patients experienced grade ≥3 treatment-related adverse events (TRAEs). The most common grade 3-4 adverse events were anaemia (46%) and neutropenia (18%) in the talazoparib arm. There were more
talazoparib/placebo dose interruptions (62% vs. 21%) and dose reductions (53% vs. 7%) due to adverse events in the talazoparib group than in the placebo group. The most common adverse events leading to discontinuation of talazoparib were anaemia (8%) and neutropenia (3%). There were no treatment-related deaths in the talazoparib arm and two deaths in the placebo group. Myelodysplastic syndrome was reported in one patient during the safety reporting period and acute myeloid leukaemia was reported in one patient during the follow-up period (both in the talazoparib arm).1
To date, there is strong evidence that the co-inhibition of AR and PARP results in therapeutic synergy.2 In the TALAPRO-2 study, talazoparib plus enzalutamide demonstrated a clinically meaningful and statistically significant improvement in rPFS versus standard of care enzalutamide for the all-comers population (the primary endpoint), HRR deficient subgroup, and HRR-non-deficient or unknown subgroup, as well as in the HRR-non-deficient subgroup by prospective tumour tissue testing only. These results thus support the use of talazoparib plus enzalutamide as a first-line treatment option in patients with mCRPC.1
REFERENCES
Article written on request of Pfizer.
240531 – August 2024
NAME OF THE MEDICINAL PRODUCT: Talzenna 0.1 mg, 0.25 mg and 1 mg hard capsules. QUALITATIVE AND QUANTITATIVE COMPOSITION: Talzenna 0.1 mg, 0.25 mg resp. 1 mg hard capsules: Each hard capsule contains talazoparib tosylate equivalent to 0.1 mg, 0.25 mg resp. 1 mg talazoparib. PHARMACEUTICAL FORM: Hard capsule (capsule). Talzenna 0.1 mg hard capsules: Opaque, approximately 14 mm × 5 mm hard capsule with a white cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.1” in black). Talzenna 0.25 mg hard capsules: Opaque, approximately 14 mm × 5 mm hard capsule with an ivory cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.25” in black). Talzenna 1 mg hard capsules: Opaque, approximately 14 mm × 5 mm hard capsule with a light red cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 1” in black). CLINICAL PARTICULARS: Therapeutic indications: Breast cancer: Talzenna is indicated as monotherapy for the treatment of adult patients with germline BRCA1/2‑mutations, who have HER2negative locally advanced or metastatic breast cancer. Patients should have been previously treated with an anthracycline and/or a taxane in the (neo)adjuvant, locally advanced or metastatic setting unless patients were not suitable for these treatments. Patients with hormone receptor (HR)‑positive breast cancer should have been treated with a prior endocrine‑based therapy, or be considered unsuitable for endocrine‑based therapy. Prostate cancer: Talzenna is indicated in combination with enzalutamide for the treatment of adult patients with metastatic castration‑resistant prostate cancer (mCRPC) in whom chemotherapy is not clinically indicated. Posology and method of administration: Treatment with Talzenna should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. Patient selection: Breast cancer: Patients should be selected for the treatment of breast cancer with Talzenna based on the presence of deleterious or suspected deleterious germline BRCA mutations determined by an experienced laboratory using a validated test method. Genetic counselling for patients with BRCA mutations should be performed according to local regulations, as applicable. Prostate cancer: There is no requirement for tumour mutation testing for selection of patients with mCRPC for treatment with Talzenna. Posology: Talzenna monotherapy (breast cancer): The recommended dose is 1 mg talazoparib once daily. Patients should be treated until disease progression or unacceptable toxicity occurs. Talzenna in combination with enzalutamide (prostate cancer): The recommended dose is 0.5 mg talazoparib in combination with 160 mg enzalutamide once daily. Patients should be treated until disease progression or unacceptable toxicity occurs. Medical castration with luteinising hormone releasing hormone (LHRH) analogue should be continued during treatment in patients not surgically castrated. Please refer to the full enzalutamide product information for the recommended posology. Missing dose: If the patient vomits or misses a dose of Talzenna, an additional dose should not be taken. The next prescribed dose should be taken at the usual time. Dose adjustments: To manage adverse drug reactions, interruption of treatment or dose reduction based on severity and clinical presentation should be considered (see Table 1). Recommended dose reduction levels for talazoparib monotherapy (breast cancer) and for talazoparib when used in combination with enzalutamide (prostate cancer) are indicated in Table 2 and Table 3, respectively. Complete blood count should be obtained prior to starting talazoparib therapy and monitored monthly and as clinically indicated (see Table 1 and section 4.4). Table 1. Dose adjustments for adverse reactions: [a] Withhold Talzenna until levels resolve to; [b] Resume Talzenna. Haemoglobin < 8 g/dL: [a] ≥ 9 g/dL; [b] Resume Talzenna at next lower dose. Platelet count < 50,000/μL: [a] ≥ 75,000/μL; [b] Resume Talzenna at next lower dose. Neutrophil count < 1 000/μL: [a] ≥ 1 500/μL; [b] Resume Talzenna at next lower dose. Non‑haematologic adverse reaction Grade 3 or Grade 4: [a] ≤ Grade 1; [b] Consider resuming Talzenna at next lower dose or discontinue. Table 2. Dose reduction levels for talazoparib monotherapy (breast cancer): Talazoparib dose level (breast cancer): Recommended starting dose: 1 mg once daily. First dose reduction: 0.75 mg once daily. Second dose reduction: 0.5 mg once daily. Third dose reduction: 0.25 mg once daily. Table 3. Dose reduction levels for talazoparib when used in combination with enzalutamide (prostate cancer): Talazoparib dose level (prostate cancer): Recommended starting dose: 0.5 mg once daily. First dose reduction: 0.35 mg once daily. Second dose reduction: 0.25 mg once daily. Third dose reduction: 0.1 mg once daily. Please refer to the full enzalutamide product information for dose adjustment for adverse reactions associated with enzalutamide. The intended use of the 0.1 mg capsule is to support dose modifications and it is not interchangeable with other strengths. Concomitant treatment with inhibitors of P-glycoprotein (P-gp): Talzenna monotherapy (breast cancer): Strong inhibitors of P‑gp may lead to increased talazoparib exposure. Concomitant use of strong P‑gp inhibitors during treatment with talazoparib should be avoided. Co‑administration should only be considered after careful evaluation of the potential benefits and risks. If co‑administration with a strong P‑gp inhibitor is unavoidable, the Talzenna dose should be reduced to the next lower dose. When the strong P‑gp inhibitor is discontinued, the Talzenna dose should be increased (after 3‑5 half‑lives of the P‑gp inhibitor) to the dose used prior to the initiation of the strong P‑gp inhibitor. Talzenna when used in combination with enzalutamide (prostate cancer): The effect of co‑administration of P‑gp inhibitors on talazoparib exposure when talazoparib is given in combination with enzalutamide has not been studied. Therefore, concomitant use of P‑gp inhibitors during treatment with talazoparib should be avoided. Special populations: Hepatic impairment: No dose adjustment is required for patients with mild hepatic impairment (total bilirubin ≤ 1 × upper limit of normal [ULN] and aspartate aminotransferase (AST) > ULN, or total bilirubin > 1.0 to 1.5 × ULN and any AST), moderate hepatic impairment (total bilirubin > 1.5 to 3.0 × ULN and any AST), or severe hepatic impairment (total bilirubin > 3.0 × ULN and any AST). Talzenna in combination with enzalutamide is not recommended for use in patients with severe hepatic impairment (Child‑Pugh classification C), as pharmacokinetics and safety have not been established in these patients. Renal impairment: Breast cancer: No dose adjustment is required for patients with mild renal impairment (60 mL/min ≤ creatinine clearance [CrCL] < 90 mL/min). For patients with moderate renal impairment (30 mL/min ≤ CrCL < 60 mL/min), the recommended starting dose of Talzenna is 0.75 mg once daily. For patients with severe renal impairment (15 mL/min ≤ CrCL < 30 mL/min), the recommended starting dose of Talzenna is 0.5 mg once daily. Talzenna has not been studied in patients with CrCL < 15 mL/min or patients requiring haemodialysis. Prostate cancer: No dose adjustment is necessary for patients with mild renal impairment (60 mL/min ≤ creatinine clearance [CrCL] < 90 mL/min). For patients with moderate renal impairment (30 mL/min ≤ CrCL < 60 mL/min), the recommended dose of Talzenna is 0.35 mg once daily in combination with enzalutamide orally once daily. For patients with severe renal impairment (15 mL/min ≤ CrCL < 30 mL/min), the recommended dose of Talzenna is 0.25 mg once daily in combination with enzalutamide once daily. Talzenna has not been studied in patients with CrCL < 15 mL/min or patients requiring haemodialysis. Elderly: No dose adjustment is necessary in elderly (≥ 65 years of age) patients. Paediatric population: The safety and efficacy of Talzenna in children and adolescents < 18 years of age have not been established. No data are available. Method of administration: Talzenna is for oral use. To avoid contact with the capsule content, the capsules should be swallowed whole, and must not be opened or dissolved. They can be taken with or without food. Contraindications: Hypersensitivity to the active substance or to any of the excipients listed. Breast‑feeding. Undesirable effects: Summary of the safety profile: The overall safety profile of Talzenna is based on pooled data from 1 088 patients, including 690 patients who received talazoparib monotherapy at 1 mg daily in clinical studies for solid tumours and 398 patients with mCRPC who received talazoparib 0.5 mg in combination with enzalutamide 160 mg in the TALAPRO‑2 study. The most common (≥ 20%) adverse reactions in patients receiving talazoparib in these clinical studies were anaemia (55.6%), fatigue (52.5%), nausea (35.8%), neutropenia (30.3%), thrombocytopenia (25.2%) and decreased appetite (21.1%). The most common (≥ 10%) Grade ≥ 3 adverse reactions of talazoparib were anaemia (39.2%), neutropenia (16.5%) and thrombocytopenia (11.1%). Dose modifications (dose reductions or dose interruptions) due to any adverse reaction occurred in 58.7% of patients receiving Talzenna 1 mg monotherapy. The most common adverse reactions leading to dose modifications were anaemia (33.5%), neutropenia (11.7%) and thrombocytopenia (9.9%). Permanent discontinuation due to an adverse reaction occurred in 2.9% of patients receiving Talzenna; the most common was anaemia (0.6%). The median duration of exposure was 5.6 months (range 0.0 to 70.2). Dose interruptions of Talzenna due to adverse reactions occurred in 62.1% of patients with mCRPC receiving Talzenna in combination with enzalutamide; the most common was anaemia (44%). Dose reductions of Talzenna due to adverse reactions occurred in 52.8% of patients; the most common was anaemia (43.2%). Permanent discontinuation of Talzenna due to adverse reactions occurred in 18.8% of patients; the most common was anaemia (8.3%). The median duration of talazoparib exposure was 86 weeks (range 0.29 to 186.14). Tabulated list of adverse reactions: Table 4 summarises adverse reactions based on pooled dataset listed by system organ class, and frequency category. Frequency categories are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10) and uncommon (≥ 1/1 000 to < 1/100). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. Table 4. Adverse reactions based on pooled dataset from 8 studies (N=1 088). Adverse reactions are mentioned for: All grades n (%), Grade 3 n (%), Grade 4 n (%). Neoplasms benign, malignant and unspecified (including cysts and polyps): Uncommon: Myelodysplastic syndrome/Acute myeloid leukaemiaa: 2 (0.2), 1 (< 0.1), 1 (< 0.1). Blood and lymphatic system disorders: Very common: Thrombocytopeniab: 274 (25.2), 88 (8.1), 33 (3.0); Anaemiac: 605 (55.6), 411 (37.8), 16 (1.5); Neutropeniad: 330 (30.3), 163 (15.0), 17 (1.6); Leukopeniae: 195 (17.9), 52 (4.8), 2 (0.2). Common: Lymphopeniaf: 88 (8.1), 37 (3.4), 4 (0.4). Metabolism and nutrition disorders: Very common: Decreased appetite: 230 (21.1), 11 (1.0), 0 (0.0). Nervous system disorders: Very common: Dizziness: 157 (14.4), 4 (0.4), 1 (< 0.1); Headache: 207 (19.0), 8 (0.7), N/A. Common: Dysgeusia: 68 (6.3), 0 (0.0), 0 (0.0). Vascular disorders: Common: Venous thromboembolism*g: 36 (3.3%), 23 (2.1%), 2 (0.2%). Gastrointestinal disorders: Very common: Vomiting: 167 (15.3), 9 (0.8), 0 (0.0); Diarrhoea: 205 (18.8), 4 (0.4), 0 (0.0); Nausea: 389 (35.8), 10 (0.9), N/A; Abdominal painh: 162 (14.9), 12 (1.1), N/A. Common: Stomatitis: 54 (5.0), 0 (0.0), 0 (0.0); Dyspepsia: 69 (6.3), 0 (0.0), N/A. Skin and subcutaneous tissue disorders: Very common: Alopecia: 189 (17.4), N/A, N/A. General disorders and administration site conditions: Very common: Fatiguei: 571 (52.5), 58 (5.3), N/A. Abbreviations: n=number of patients; N/A=not applicable. * Grade 5 adverse reactions were reported. a. See also section 4.4. b. Includes preferred terms of thrombocytopenia and platelet count decreased. c. Includes preferred terms of anaemia, haematocrit decreased, haemoglobin decreased and red blood cell count decreased. d. Includes preferred terms of neutropenia and neutrophil count decreased. e. Includes preferred terms of leukopenia and white blood cell count decreased. f. Includes preferred terms of lymphocyte count decreased and lymphopenia. g. Includes preferred terms of pulmonary embolism, deep vein thrombosis, embolism venous and venous thrombosis. See also section 4.4. h. Includes preferred terms of abdominal pain, abdominal pain upper, abdominal discomfort and abdominal pain lower. i. Includes preferred terms of fatigue and asthenia. Description of selected adverse reactions: Myelosuppression: Myelosuppression-related adverse reactions of anaemia, neutropenia and thrombocytopenia were very commonly reported in patients treated with talazoparib. Grade 3 and Grade 4 myelosuppression‑related events were reported for anaemia in 37.8% and 1.5% of patients, neutropenia in 15.0% and 1.6%, and thrombocytopenia in 8.1% and 3.0%. No deaths were reported due to myelosuppression‑related adverse reactions. In monotherapy studies (1 mg/day population), the most frequent myelosuppression‑related adverse events associated with dose modifications were anaemia (33.5%), neutropenia (11.7%) and thrombocytopenia (9.9%) reported for up to approximately 30% of patients in the talazoparib 1 mg/day population and the one associated with permanent study drug discontinuation was anaemia reported in 0.6% of patients. In patients with mCRPC treated with talazoparib in combination with enzalutamide, anaemia led to talazoparib dose interruption in 44.0% of patients, decreased neutrophil count in 13.6%, and decreased platelet count in 7.8%. Overall, 42.5% of patients required blood transfusions. The most common blood transfusion was of packed red blood cells 39.2%. Discontinuation due to anaemia, neutropenia and thrombocytopenia occurred, respectively, in 8.3%, 3.3% and 0.5% of patients. Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Federal Agency for Drugs and Health Products – Vigilance Department, Galileelaan – Avenue Galilée 5/03, 1210 Brussels (website: www.eenbijwerkingmelden.be – www.notifieruneffetindesirable.be; e-mail: adr@fagg-afmps.be). MARKETING AUTHORISATION HOLDER: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium. MARKETING AUTHORISATION NUMBER(S) : Talzenna 0.1 mg hard capsules: EU/1/19/1377/007. Talzenna 0.25 mg hard capsules: EU/1/19/1377/001, 002, 003, 004. Talzenna 1 mg hard capsules: EU/1/19/1377/005, 006. DELIVERY: On medical prescription. DATE OF REVISION OF THE TEXT: 04/2024. Detailed information on this medicinal product is available on the website of the European Medicines Agency https://www.ema.europa.eu.