Hypoxic respiratory failure from acute respiratory distress syndrome (ARDS) is the leading cause of mortality in patients with Coronavirus disease 2019 (COVID-19). Emerging evidence suggests that endothelial cells are essential contributors to the initiation and propagation of severe COVID-19 disease. This study discusses the current insights into the link between endothelial cells, viral infection, and inflammatory changes and proposes novel therapeutic strategies.
Hypoxic respiratory failure from acute respiratory distress syndrome (ARDS) is the leading cause of mortality in patients with Coronavirus disease 2019 (COVID-19). Emerging evidence suggests that pulmonary endothelial cells (ECs) contribute to the initiation and propagation of ARDS by altering vessel barrier integrity, promoting a pro-coagulative state, inducing vascular inflammation (endotheliitis) and mediating inflammatory cell infiltration. The endothelium maintains vascular integrity and barrier function. It prevents inflammation by limiting endothelial cell (EC)-immune cell and EC-platelet interaction and inhibits coagulation by expressing coagulation inhibitors, blood clot-lysing enzymes and a glycocalyx with anti-coagulation properties. Lung ECs are enriched in transcriptomic signatures indicating immunoregulation, suggesting a role in immune surveillance against respiratory pathogens.
Around 30% of hospitalised patients with COVID-19 develop progressive lung damage, partially due to an overreacting inflammatory response (Figure 1). Vascular leakage and pulmonary oedema are caused by multiple mechanisms. First, the virus can directly infect the EC cells, producing endotheliitis and eventually dysfunction, lysis and death. Second, the virus can also bind to the ACE2 receptor, which impairs the activity of ACE2, resulting in increased vascular permeability. Third, the neutrophils recruited to the pulmonary ECS produce histotoxic mediators, including ROS. Fourth, immune cells, inflammatory cytokines and vasoactive molecules lead to enhanced EC contractility and the loosening of inter-endothelial junctions, leading to inter-endothelial gaps. Finally, the cytokines IL-1β and TNF activate glucuronidases that degrade the glycocalyx but also upregulate hyaluronic acid synthase 2, leading to increased deposition of hyaluronic acid in the extracellular matrix and promoting fluid retention.
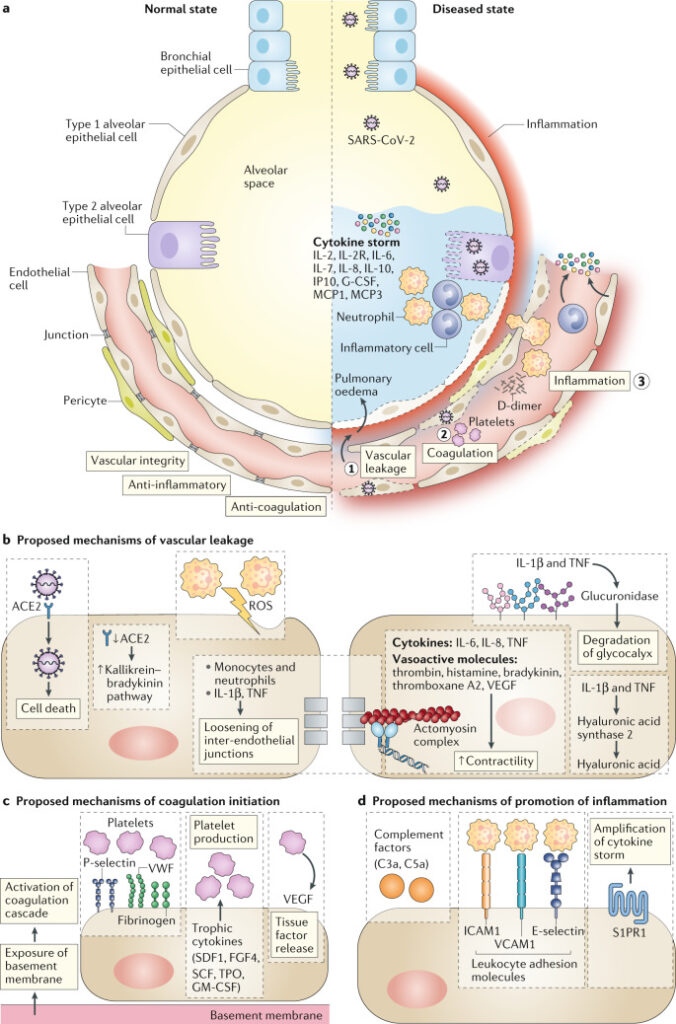
Figure 1. Proposed vessel–lung tissue interface in normal state and in COVID-19 disease.
On the left, the normal interface between the alveolar space and endothelial cells. On the right side, highlights pathophysiological features of coronavirus disease 2019 (COVID-19) in the lung, including loss of vascular integrity (1), activation of the coagulation pathway (2) and inflammation (3). b–d | Proposed contributing endothelial cell-specific mechanisms are detailed. ROS, reactive oxygen species; S1PR1, sphingosine 1 phosphate receptor 1; VWF, von Willebrand factor.
Disruption of vascular integrity and EC death leads to exposure of the thrombogenic basement membrane, which results in the activation of the clotting cascade. ECs activated by IL-1β and TNF also initiate coagulation by expressing P- selectin, von Willebrand factor and fibrinogen, to which platelets bind. In turn, ECs release trophic cytokines that further augment platelet production. Platelets also release VEGF, which triggers ECs to upregulate the expression of tissue factor, the prime activator of the coagulation cascade. As a result of the DIC and clogging/congestion of the small capillaries by inflammatory cells, as well as possible thrombosis in larger vessels, lung tissue ischaemia develops, which triggers angiogenesis and potential EC hyperplasia.
Many patients with severe COVID-19 show signs of a cytokine storm, which leads to further EC dysfunction, DIC, inflammation and vasodilation. This results in alveolar dysfunction, ARDS with hypoxic respiratory failure and ultimately multi-organ failure and death. EC dysfunction likely co-determine this uncontrolled immune response, by promoting inflammation by expressing leukocyte adhesion molecules, thereby facilitating the accumulation and extravasation of leukocytes, including neutrophils, which enhance tissue damage. The denudation of the pulmonary vasculature could lead to activation of the complement system, promoting the accumulation of neutrophils and pro-inflammatory monocytes that enhance the cytokine storm. In influenza infection, modulation of S1PR1 in pulmonary ECs dampens the cytokine storm, and something similar could occur during COVID-19 infection. The excessive lymphopenia could relate to the recruitment of lymphocytes away from the blood by activated lung ECs.
Additional evidence suggests a link between ECs, pericytes and COVID-19. First, risk factors for COVID-19 are characterised by altered EC metabolism. Second, a few reports suggest an increased incidence of Kawasaki disease, a vasculitis, in young children with COVID-19. Third, severe COVID-19 is characterised by multi-organ failure, raising the question of how and to what extent the damaged pulmonary endothelium no longer offers a barrier to viral spread from the primary infection site.
The proposed central role of ECs in COVID-19 disease escalation prompts the question whether vascular normalisation strategies during the maladapted immune response could be useful. Although further studies need to validate the hypothesis, the possibility that ECs and other vascular cells are important players paves the way for future therapeutic opportunities.
Reference